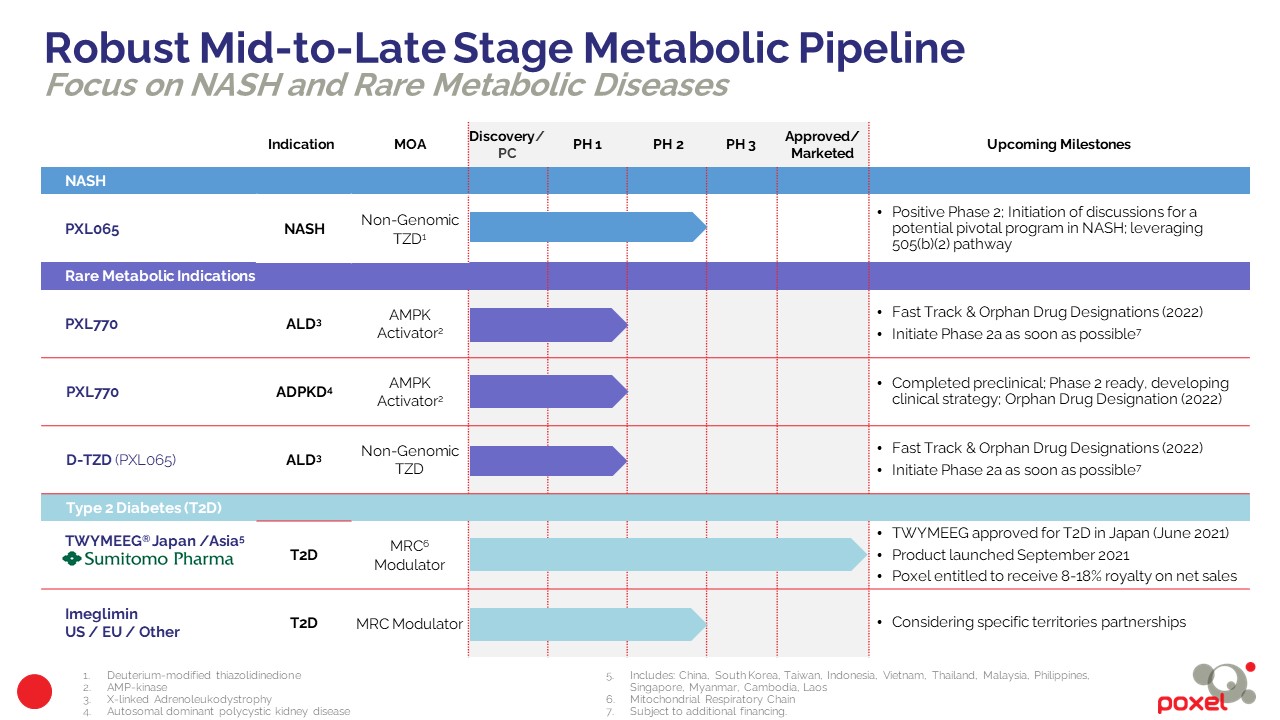
NASH
PXL065
PXL065 is deuterium-modified R-pioglitazone, currently in a streamlined Phase 2 trial (DESTINY-1). Although pioglitazone is not approved by the FDA for the treatment of NASH, it is the most extensively studied drug for NASH and has demonstrated NASH resolution and improvements in fibrosis in multiple clinical trials. Pioglitazone is the only drug recommended for biopsy-proven NASH patients by the Practice Guidelines published by the American Association for the Study of Liver Diseases (AASLD) and the European Association for the Study of the Liver (EASL). Pioglitazone’s off-label use for NASH, however, has been limited due to the PPARγ-related side effects, which include weight gain, bone fractures and fluid retention.
Pioglitazone is a 1:1 mixture of two mirror-image compounds (R- and S-stereoisomers) that interconvert in vivo. Using deuterium, we stabilized each stereoisomer and characterized their different pharmacological properties. PXL065 is the R-pioglitazone isomer stabilized by deuterium. In in vitro studies, PXL065 has been shown to lack PPARγ activity while retaining non-genomic TZD actions. In preclinical animal models, PXL065 exhibits similar anti-inflammatory and NASH activity as pioglitazone with little or no weight gain or fluid retention, side effects which are associated with the S-stereoisomer.
NASH Phase 2 (DESTINY 1 trial) results available to-date show statistically significant effects of PXL065 on liver fat content, biomarkers related to liver fibrogenesis-fibrosis risk, as well as positive effects on fibrosis and other key parameters based on histology analysis. Relative to published data for pioglitazone, reduced potential for weight gain and edema was also evident. Based upon preclinical, Phase 1 and Phase 2 results, Poxel believes that PXL065 may have a better therapeutic profile than pioglitazone for NASH and may also have suitable properties for further development in other indications including adrenoleukodystrophy (ALD).
NASH
Rare Diseases
PXL770 and D-TZD platform (PXL065) in Adrenoleukodystrophy (ALD)
PXL770 is a first-in-class direct AMPK activator. Clinical Phase 1 and 2a development to-date has demonstrated target engagement and translation of several metabolic efficacy parameters to humans which suggests the likelihood of broader translation for this mechanism. PXL770 was observed to ameliorate key features of ALD in preclinical models, including both patient-derived cells and a classical rodent disease model.
PXL065 was observed to ameliorate key features of ALD in preclinical models, including both patient-derived cells and a classical rodent disease model. PXL065 has open IND for ALD in the U.S. Preclinical and clinical results obtained with pioglitazone and a related molecule, leriglitazone (Minoryx), support the potential utility of PXL065 in ALD. PXL065 differentiates from pioglitazone and leriglitazone in several ways including its potential for greater efficacy and reduced side effects.
A Phase 2a clinical POC biomarker studies of PXL770 is planned to initiate as soon as possible, subject to financing. The initial focus will be on ALD patients with adrenomyeloneuropathy (AMN), the largest subtype of ALD. Each study will enroll adult male AMN patients and assess the effect of PXL065 over 12 weeks of treatment on pharmacokinetics, safety, and efficacy using relevant biomarkers, including the impact on elevated very long-chain fatty acids (VLCFA), the hallmark plasma marker of disease. Considering the DESTINY-1 results for PXL065 in NASH, which validated the deuterium-modified thiazolidinedione (TZD) platform, a second identical study is planned in order to assess the potential of the deuterium-modified TZD platform with PXL065 in ALD.
In early 2022, the U.S. Food and Drug Administration (FDA) granted Fast Track and Orphan Drug Designation to PXL065 and PXL770 for the treatment of patients with adrenomyeloneuropathy (AMN), the most common form of X-linked adrenoleukodystrophy (ALD). The Fast Track Designation is designed to expedite development of pharmaceutical products which demonstrate the potential to address unmet medical needs in serious or life-threatening conditions. Orphan Drug Designation is granted by the FDA to novel therapeutics for diseases or conditions that affect fewer than 200,000 individuals in the U.S. Orphan Drug Designation gives a company a potential seven-year window of exclusive marketing rights following FDA approval, along with a reduction in certain application fees, and tax credits for expenses related to qualified clinical trials conducted after orphan designation is received.
PXL770 in Autosomal-Dominant Polycystic Kidney Disease (ADPKD)
The pathophysiology of ADPKD involves altered kidney metabolism and there is a strong rationale for AMPK activation as a therapeutic approach. Having completed the preclinical assessment of PXL770 in ADPKD demonstrating compelling benefits of strong suppression of cyst formation in vitro and in vivo, PXL770 is a Phase 2 ready program. In H2 2022, the U.S. Food and Drug Administration (FDA) granted Orphan Drug Designation to PXL770 for the treatment of patients with ADPKD.
Rare Diseases
Diabetes
Imeglimin is a first-in-class novel medicine that is now approved and commercialized as TWYMEEG in Japan for the treatment of Type 2 diabetes. It has a dual mode of action which corrects hyperglycemia by improving insulin secretion in response to glucose from pancreatic b-cells and ameliorating insulin resistance in key tissues (muscle and liver). Imeglimin also has the potential to exert cardiorenal disease modifying benefits and protective effects on pancreatic islet b-cell survival. This unique profile supports Imeglimin's use for the treatment of Type 2 diabetes in almost all stages of the current treatment paradigm.
Japan/Asia
In Japan, China, Taiwan, South Korea and 9 other Southeast Asian countries, Sumitomo Pharma is our strategic partner for Imeglimin. A new drug application for TWYMEEG Tablets 500mg1 (International Nonproprietary Name (INN): Imeglimin hydrochloride), for the treatment of type 2 diabetes, was approved in Japan in June 2021. Japan is the first country in the world to approve Imeglimin. Sumitomo Pharma, the diabetes market leader in Japan, launched TWYMEEG in September 2021.
The TWYMEEG approval in Japan is supported by numerous preclinical and clinical studies, including the Phase 3 TIMES (Trials of IMeglimin for Efficacy and Safety) program managed jointly by Poxel and Sumitomo Pharma, which included three pivotal trials to evaluate TWYMEEG’s efficacy and safety in over 1,100 patients. In all three trials, TWYMEEG met its primary endpoints and objectives and was observed to exhibit a favorable safety and tolerability profile when used alone or in combination with standard of care.
In China, Taiwan, South Korea, Indonesia, Vietnam, Thailand, Malaysia, the Philippines, Singapore, Myanmar, Cambodia, and Laos, Sumitomo Pharma will be solely responsible for the development and commercialization of Imeglimin.
1Dosage and administration: In general, for adults, 1,000 mg of Imeglimin hydrochloride is orally administered twice daily in the morning and evening.
U.S. and Europe
Poxel owns full rights for the US, Europe, and the other countries not covered by the partnership with Sumitomo Pharma. The Company is considering opportunities to leverage the Imeglimin data package in specific territories, including those resulting from inbound interest.
Diabetes
Additional Pipeline Opportunities
Preclinical studies are ongoing to evaluate our adenosine monophosphate-activated protein kinase (AMPK) activation and dTZD platforms in additional chronic and rare orphan metabolic diseases.